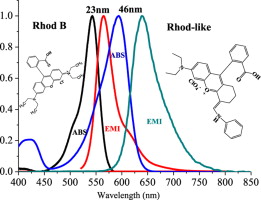
Energy transfer occurs between pairs of molecules, often called FRET pairs. Perhaps the most studied is rhodamine and fluorescein. We will use uranine, which is simply water soluble fluorescein.
The laboratory protocol is a generic for testing optical properties of fluorophores. We will use the combination of Time Correlated Single Photon Counting (TCSPC) for the time-resolved measurement of tauobs. We know that fluorescein and rhodamine are probably not an optimal FRET pair when excited by 509 nm laser LED. We also have a 390 nm laser.
A review of fluorescence spectroscopy including topics such as mirror-image relationship and how to think about the symmetries of modes coupled to allowed transitions. All modes should be totally symmetric. It is very important to spectroscopy that there are exceptions, but we must understand that non-totally symmetric modes are often coupled to weak processes that are made allowed by the distortion of the molecule away from its point group symmetry.
The detailed explanation for the line shape resides in the probability for transitions at each geometry.
Empirical methods for calculating the spectral overlap integral
We can use spectral data from databases such as Photochem CAD. The presentation describes how to think about donor-acceptor overlaps using sepctral data. If data from Photochem CAD are used they must be interpolated to make all data files compatible. This is necessary for determination of spectral integrals.
Photochem CAD was developed in the Lindsey lab at NCSU as a tool for comparing spectrocopic properties of molecules with strong emission. The explanation takes a student through the steps involved in spreadsheet or computer calculation of spectral overlap.
How does one calculate the overlaps of these functions in a general case between two different molecules? To answer this question we first must understand line shapes of quantum transitions. The strongly allowed transitions are coupled only to totally-symmetric modes. The theory that calculates the overlaps under these conditions is discussed in the link below.
Now that you can see how it is done for fluorescein (uranine) and rhodamine 6G, you can try it yourself for the rhodamine 6G - sulforhodamine 101 FRET pair.
Interpolated absorption and emission data from Photochem CAD
Thus far, spectra and overlaps have been calculated using recorded spectra from Photochem CAD. One important point is that the spectra were measured in ethanol. There could be a difference in water. The most important of the empirical method is that you can apply it to your own data.
First principles calculation of the spectral overlap integral
The above applications are empirical in the sense that we have used the experimental data to model the spectral overlap integral. A deeper level of analysis uses a first principles calculation of the line shape for both donor emission and acceptor absorption spectra, rather than the experimental data. Of course, we must compare these calculated values to the experimental data.
The mirror-image relationship of fluorescence spectra is also useful predicting energy transfer probability. The energy transfer rate constant is proportional to the overlap of the donor emission and acceptor absorption. This section discusses spectral modeling, which gives a method for calculating those line shapes from first principles.
The only required input is the Raman spectrum. Recalling the Raman lines resonant with strongly allowed transitions are totally symmetric and Franck-Condon allowed we can use sum-over-states or wavepacket methods to calculate the line shape.
Studies of other molecule excitation and emission spectra are similarly helped by studying the Raman active modes. These modes are the totally symmetric modes and they provide us with a simple model for calculation of the absorption spectrum.
Using SecureShellClient 3.2.9 log in to the HPC (login.hpc.ncsu.edu).
Using a FileTransfer Window in SecureShellClient 3.2.9 transfer ET.tar to the HPC.
Check that ET.tar is on the HPC. Use: $ ls
Unpack ET.tar using the "tar" command:
$ tar -xvf ET.tar
Sample input files and executables "raman_spec" and "timetherm" appear in the directory. You may download or inspect these files here as well.
A recommended number of modes for this analysis is 7, but the program can handle larger numbers of modes if needed. The reasoning is that large aromatic molecules typically have coupling to ring modes upon absorption. These ring modes range from 1100-1700 cm-1 and are typically quite weak, S << 0.1.
The timetherm input file contains the same wavenumbers and electron-vibration coupling as in the model spectrum, but also has a number of the control parameters. You can see what each parameter is by typing $ timetherm and putting in sample input by hand.
We can distinguish between the intensity of absorption and fluorescence, which are interrelated by the Planck-Einstein analysis. The absorption line shapes can be calculated using the wave packet formalism in the program timetherm.
|