Protein folding
Protein
folding is a relatively complex process because the linear peptide chain has
order that leads to folding of specific amino acids on the interior and others
on the exterior of the folded protein.
We can examine the contribution to the free energy of the various amino acid
side chains and secondary structure elements in order to help determine the
rules for protein folding. While this is
long from a solved problem, much progress has been made
in the understanding of the energetic and kinetic contributions made by
specific structures. First, we consider
the globar problem of protein folding. Subsequently, we will decompose the problem
into its component parts by considering, hydrogen bonding, electrostatic, and
hydrophobic interactions.
The
global consideration for protein folding starts with the consideration of a
two-state protein folding model. While this is probably a simplification for
most protein folding problems, the existing experimental data are consistent
with a two-state model in the majority of cases. Therefore, we are justified in using it. It is certainly the most appropriate model to
explain the issues in protein folding.
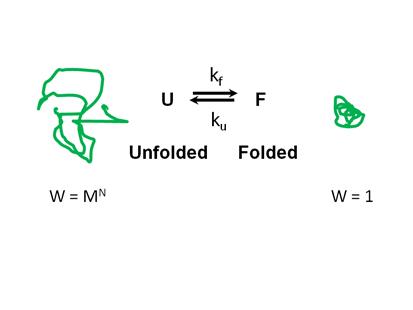
Figure
15.1. Depiction of the two-state model for protein folding.
The two-state model implies that an
equilibrium exists between the folded and unfolded conformations. The equilibrium constant is:
![]()
(1)
However, we often measure the
relative amount that is folded, i.e. the fraction of folded protein, ff. Of course, the fraction of unfolded protein fu is just 1 – ff. Therefore, we can also write the equilibrium
constant as
![]()
(2)
Solving for ff
we obtain
![]()
(3)
We can use our knowledge of the
relationship of K and DGo to
write
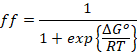
(4)
which can
also be expressed as
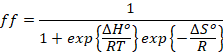
(5)
The temperature at which the
protein is 50% folded can be defined as Tm
the melt temperature.
At Tm
, DGo = 0 or Tm
= DHo/DSo.
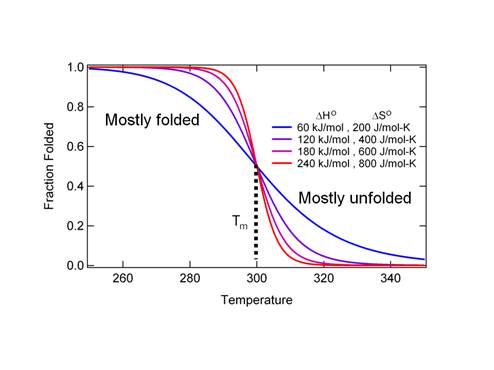
Figure
2. An equilibrium melt curve. Tm
is called the melt temperature.
It is the temperature at which the protein begins to unfold.
Figure 2 shows a compensation of DHo and DSo such that the Tm is 300 K in all
cases. The greater DHo and DSo the steeper the curve. Most realistic cases have relatively small
values of
DHo
so that the curves are shallow (see the blue curve). A typical protein contains a few
salt-bridges, several hundred hydrogen bonds and several thousand van der Waals
interactions. In spite of all these interactions
proteins are only marginally stable. Typical
DG values for folding of proteins are
in the range of -5 to -15 kcal/mol
i.e. not much greater than the energy of 2 or 3 hydrogen bonds. This is because
of several effects which cancel each other out. The
enthalpy change of protein folding (DH)
is dominated by hydrogen bonds. In the unfolded state the polar groups of the protein will H-bond to solvent
molecules and in the folded state these polar groups will H-bond with each
other. Hence the overall enthalpy change on folding is
small. The hydrophobic effect is thought to make the
largest contribution to DG that
stabilizes the folded state. The hydrophobic effect attributes the poor
solubility of non-polar groups in water to the ordering of the surrounding
water molecules causing them to form an ice-like cluster. This is shown in
Figure 3.
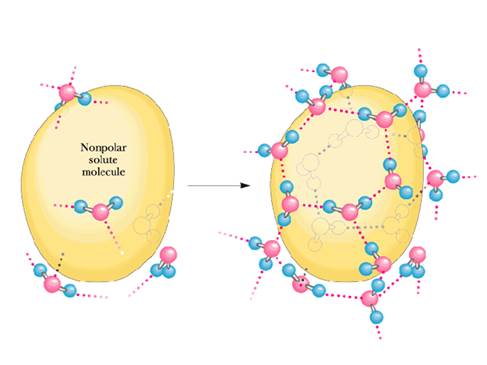
Figure 3. The hydrophobic
effect. The left panel shows the
unfavorable interactions of water with a hydrophobic object. O-H interacts strongly with oxygen lone
pairs, but not C-H groups on hydrophobic molecules. The right panel shows water with an organized
structure around a hydrophobic solute.
The water molecules have rotated to avoid contact of O-H groups with the
solute.
On the other hand, the
conformational (or configurational) entropy is the largest destabilizing
force. The number of possible
conformations in the unfolded state is large.
We have shown in a previous chapter that the number of accessible states
(conformations) W = MN where M is the number of conformers for each
monomer and N is the number of monomers in the biopolymer. In the folded state there is a relatively small number of
conformations. In an ideal folded
protein W = 1, i.e. there is a unique
conformation. In that case
the conformational entropy can be estimated to be
![]()
(6)
Hence,
![]()
(7)
which is a
significant contribution. In fact, the
entropy issue is a major issue in protein folding. It was first raised in 1968
by Levinthal in a famous article that
discussed the consequences of the statistical nature of protein
conformations. Assuming that the protein
searches randomly for the correct conformation, the number of states the must
be sampled is enormous. For a protein
like human myoglobin with 160 amino acids and an average of 6 conformations per
side chain, the total number of conformations is W = 6160 ![]() 5 x 10116! The kinetics of this entropic contribution
presents a paradox, known as the Levinthal
paradox. Even with a
ultra-rapid random search it would take longer than the age of the universe to
find the folded conformation. Is the
large negative entropy overcome in a single step or is protein folding a more
gradual and complex process. We will
revisit this question when we talk about kinetics. However, at this point it is clear that
conformational (statistical) entropy is the dominant force that leads to
protein unfolding. Moreover, since it is
an entropy, it is temperature dependent.
5 x 10116! The kinetics of this entropic contribution
presents a paradox, known as the Levinthal
paradox. Even with a
ultra-rapid random search it would take longer than the age of the universe to
find the folded conformation. Is the
large negative entropy overcome in a single step or is protein folding a more
gradual and complex process. We will
revisit this question when we talk about kinetics. However, at this point it is clear that
conformational (statistical) entropy is the dominant force that leads to
protein unfolding. Moreover, since it is
an entropy, it is temperature dependent.
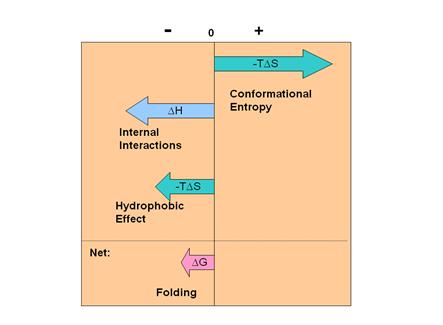
Figure
4. Summary of contributions to the free energy of protein folding.
Figure
4 shows that the balance of the various contributions
to DGo leads to a near
cancellation such that DGo
is quite small. Under physiological
conditions DfoldingGo
< 0 for proteins that fold in the cell.
However, DGo
decreases in magnitude as the temperature increases. When DGo
= 0 the protein is in equilibrium with its unfolded
state. We often call this the “melt”
temperature, Tm, as shown in Figure 2. Above this temperature
proteins unfold or denature. Protein
unfolding is important in disease. For
example, when you run a temperature, your elevated body temperature can cause
viral or bacterial proteins to unfold.
This compromises the ability of the invader to further
infect the body. Protein folding
can also be harmful as in the case of Alzheimer’s disease, which is caused by
plaques that form with the Ab peptide
aggregates in the vicinity of a nerve cell.